
Automated or Computerized Systems
Automated or computerized systems are powerful business tools selected to meet business needs for such tasks as research and development, production, and maintenance. These tools can also generate and store evidence for meeting regulatory requirements. As in most industries, automated systems have fostered operational simplicity, efficiency, reliability, repeatability, and accuracy in all business areas from procurement to human resources. Automated systems can increase safety in the monitoring and control of risky processes and regulate processes through real-time feedback.
Regulatory agencies recognize that these systems, under certain conditions, are reliable tools that can provide benefits for all stakeholders. 21 CFR 211.68 and ICH Q7 (Section 5.4) both specifically address the conditions under which automation is considered acceptable. Good automated manufacturing practices (GAMP) 5 includes information about automation.
Automated or computerized systems, regardless of whether they are embedded in equipment or products (such as medical devices) or exist as support systems, are company assets that should be subject to all of the scrutiny and control that would be expected with any asset. The pharmaceutical professional will not necessarily be expected to know the inner workings of a computerized system, but he/she should know the characteristics of system evolution, maintenance, and regulatory compliant use.
SYSTEM LIFE CYCLE
All systems evolve through a predictable life cycle (Figure 34.1). Understanding where a system is in its life cycle is important in knowing how it got there and where it is going. The life cycle is a road map with typical characteristics. A number of phases and expectations exist that the pharmaceutical professional should expect when dealing with automated or computerized systems:

• Identifying the need. Will an automated or computerized system provide the best solution to the business problem?
• Proposing solutions. Solutions can be technical (that is, the computer system is expected to do it all, perhaps without any human intervention), procedural (that is, people will do it all by following a procedure), or, as is most common, a blend of the technical and procedural approaches where procedural steps are supported by the computer system with some human oversight.
• Deciding how to develop the system with its business use as a major goal. Should the effort be to build (that is, develop a specific program using in-house or vendor resources), buy (that is, use a program, usually a commercial product that will meet the need), or modify (that is, buy an existing product and customize it to meet the need)?
• Installing the system. Have we correctly anticipated the infrastructure and supporting systems needed? How will we replace the old system? How disruptive will it be to ongoing activities?
• Testing and acceptance. How much should we test and at what level of detail? Do the test results unequivocally and thoroughly demonstrate that the system can reliably meet the business needs?
• Training. Now that the system has been shown to be functionally appropriate, do the people who need to use it know how to use it? Will it be as useful as expected?
• Change control. All systems change, and the changes must be controlled. Systems constantly undergo three types of changes: corrective, perfective, and adaptive. Is there a clearly defined and thorough (that is, proceduralized and documented) methodology for guiding a system from its current state (the as-is) to its desired state (the to-be)?
• Backup and disaster recovery. Although hardware and software failures are becoming less frequent, they do happen. All systems bring this inherent risk. Are backup and recovery systems in place so that potential data loss is minimized? Data loss should generally be considered unacceptable, but loss of good manufacturing practices (GMP) data should be considered a failure to meet regulatory requirements.
• Decommissioning. The system will eventually become outdated, hardware and supporting systems will change, or a better way of meeting the changing business need will be found. The system may become obsolete, but the data might, for regulatory, legal, or historical reasons, need to be protected and retained. Consideration must be given to addressing the need to replace the system and maintaining (to some working degree) the old system. Not all information and processes can be easily transferred to a new system. Not all new systems can handle existing data formats and processes.
• Records. Keeping documented evidence is a regulatory must, and good business practice, but it is not practical to keep everything. Legal considerations, as well as direction from regulatory agencies, should guide what records are kept and for how long.
VALIDATION AND QUALIFICATION
Two important terms associated with but not exclusive to automated systems need to be understood: validation and qualification. In essence, these activities both meet the need for proving that a system is reliable and does what it claims to do, before and during its use, in its intended environment. Validation comprises identifying all aspects of testing that need to be completed before a product or process can be deployed for business use. It is common to write a validation master plan (VMP) to document the intent, scope, resources, schedules, activities, documentation, test types, and acceptance criteria required to achieve a validated state. Validation plans may address training and methods of change control to be used to maintain a validated state. Software and components will change, but the essential business goals regarding safety, quality, and regulatory compliance must always be met. The VMP provides guidance in achieving and maintaining qualified facilities and validated processes.
Qualification contributes to determining when a system is reliable and stable enough to support validated processes. Typical qualification phases include:
• Design qualification (DQ) is conducted to assure that the system is designed to do what is intended. This phase consists of documenting such topics as user requirements (functional and operational specifications) and includes identifying critical components, which are those features that directly impact product and process characteristics such as safety and quality. Critical components can include power supplies, equipment safety features, temperatures, pressures, weights, and alarm features. These features are implemented design features that must remain in control for the process to act predictably. In systems of great complexity where not every possible logic avenue can be practically checked, or every combination of events predicted, designing the system such that critical components or parameters are easily tested and monitored is crucial. If everything can not be tested, at least test what is critical to the safety and quality of the process or product. The design should facilitate this goal.
• Installation qualification (IQ) comprises activities for ensuring that the system is installed safely and that it will function in the environment in which it is intended. IQ, relative to automated systems, includes establishing a complete inventory of components (that is, model numbers, serial numbers, locations, part numbers, revision numbers), making sure that the components are correctly installed (that is, reliable power, correct wiring, proper grounding, working interfaces), making sure that installations are in compliance with vendor recommendations, and doing nominal checks to make sure that information is flowing through the system as expected in the design specifications.
• Operational qualification (OQ) proves that the design, its implementation, and supporting components will operate reliably together in a way that will meet the business need. OQ tests the system as it is intended to be used.
• Component qualification (CQ) (manufacturing to the correct design criteria) and
performance qualification (PQ) (consistent performance over time) are other qualification types that are sometimes needed on a specific project. DQ, IQ, and OQ are commonly used for software-intensive systems. IQ, OQ, and PQ are commonly used for equipment. The series DQ, IQ, OQ, PQ might be used for a system that includes both software and equipment. Qualification expectations and acceptance should be documented in a plan.
Addressing these needs will lead a system to be validated (it does or can do what it claims to do) or qualified (it does what, in a business sense, it is needed or expected to do). If a system has been validated and it needs to change, documented evidence must be produced that confirms that the system still performs as effectively as it did when it was originally validated, in spite of the modification.
REGULATORY REFERENCES
Pertinent regulatory references include 21 CFR 211.68, “Automated System Requirements,” and ICH Q7, GMP for API. Numerous references can be found concerning written records. 21 CFR 11 does set rules for providing written records in electronic form. In the past, record keeping relied on human observation and interpretation to supply information about which decisions would be made. The constantly improving capabilities, efficiency, and reliability of automated and computerized systems has allowed many industries, including the pharmaceutical industry, to confidently move from paper records (that were subject to human error) to electronic records (that provide greater technical accuracy).
The current regulations are clear and specific concerning what written records the United States Food and Drug Administration (FDA) needs from pharmaceutical manufacturers. In an effort to take advantage of technological improvements, provide improved consistency, and reduce paperwork, industry leaders asked FDA to set standards by which electronic records and signatures would be acceptable in meeting government regulations for written records. Federal regulations, designated 21 CFR 11, were formulated in response to this request and became law in March 1997. The discussion of electronic records is covered in the section addressing closed and open systems. Electronic signatures are acceptable in meeting regulatory needs if the signatures have the specific characteristics cited in 21 CFR 11.
OPEN AND CLOSED SYSTEMS
A few definitions exist for what constitutes open and closed computerized systems, but both systems are centered on who has access to the system and who controls the content. Open and closed refer to who has jurisdiction over the code (as in open source or closed source systems). Commercial products (commercial off-the-shelf [COTS] software) are usually written with proprietary restrictions and are closed systems; the user can not alter the basic code (or should not, as it will invalidate the warranty and infringe on copyrights). If, however, a computer system is developed in-house (or perhaps through a vendor), the pharmaceutical company will want access to the code so that modifications can be made. When purchasing a custom-developed product from a vendor (also known as a bespoke system), it is important to make sure that the agreement ensures that the purchaser will be the recipient of the code. Open systems can become closed systems if the correct restrictions are imposed.
In the pharmaceutical world, open and closed systems are distinctly defined when it comes to using electronic records and electronic signatures to meet regulatory needs. 21 CFR 11, “Electronic Records; Electronic Signatures,” provides definitions for closed and open systems. Closed systems are usually local, proprietary workplace type systems that handle unique business needs. These can be systems (that is, automated systems) that operate specific equipment or are timekeeping systems, data gathering systems, or monitoring systems. This information is important to the business and is protected from the outside world. The Internet is an open system; few restrictions are placed on access, and very little checking is done to ensure the integrity (or veracity) of the electronic records. Open systems are public-access systems. Thus, in general, closed and open systems are at opposite ends of the spectrum. Closed systems usually require the user to be granted specific access to the system (usually after some type of background check) and often require a unique log-in and password. Closed systems are strictly controlled. Open systems are for public use. If, in the pharmaceutical industry, a company would like to keep and submit electronic records to meet regulatory needs, a closed system is suggested.
CHANGE MANAGEMENT
All systems change, and pharmaceutical professionals should be familiar with the characteristics of proper change management. Regulatory agencies refer to systems as being either in control or out of control. Keeping a system in control can be tedious and painstaking, but allowing a system to drift out of control can be fatal to a business. Systems that are prone to unauthorized and uncontrolled modification are a huge risk from both safety and regulatory views. Simply put, once a system is proven to produce an acceptable product, any change to the system is presumed to change the product. Based on that concept, any modification to a system is assumed to produce a modified (that is, adulterated) product. It is crucial that any change to an automated or computerized system be accurately proposed, and reviewed for its potential impact on the product. Various methods can keep a system, and by extension the product, in control. The way that a system is kept in control is through change management. Change management has two elements: change control and configuration management.
Change control is the methodology that defines and reviews changes. Change control evolves during a system’s life cycle as the system evolves from the conceptual to productive phases. Basically, change control should not be too restrictive during the development phase, and should be rigorous during the deployment phase. The closer a system gets to the product, the more change control accountability must be demanded. It is acceptable for a developer to try things in the laboratory and to debug an evolving system to make it function, but it is not acceptable to adopt that philosophy in a system that is producing drugs for marketing. A change control methodology should, at a minimum, document:
• Who. Who needs the change? If the change is needed because of a malfunction, who witnessed or found the malfunction? Stakeholders, technical reviewers, approvers, task assignees, customers/patients, quality assurance (QA), and so on, are involved with this category.
• What. Describe the issues, problems, and enhancements, and what tasks need to be completed to resolve them. What items need to be modified to maintain control? What has been done?
• When. When did this impact the process/product, and when does it need to be fixed? If this issue has been ongoing, when did it begin, and how much product was produced during this time? In this situation, follow-up investigations are needed to determine how much product may have been affected.
• Where. What physical locations need to change? Is it a local problem, or does this have regional or global impact? If it is a functional change, where else is this function used?
• How. How does this impact the system/process/product? To what degree (that is, how severely), and how are the issues resolved?
• Why. Why does this affect the process or product? Why did it occur (that is, cause analysis)?
Configuration management is a discipline that can be thought of as software accounting. Configuration management collects and provides information about software characteristics over time and provides for protection of entities in terms of backups, vaulting, and archiving. Critical configuration management information includes:
• Identification. Configuration item identifiers (for example, part numbers, revisions) must be unique, and their characteristics clearly must be defined. The methods used to maintain the unique identification are referred to as version control. The goal is to ensure that a version comprises a unique set of characteristics. If modifications are made to version 3, it is no longer version 3; it needs another unique identifier (based on the chosen version control scheme).
• Control. Controlled configuration items must be beyond compromise either physically or electronically, and this goal can be accomplished by physical vaulting (for example, safes, limited access rooms) or by establishing electronic storage areas with limited access. Typically, electronic storage is divided into working and controlled areas. A controlled check-out/check-in scheme is recommended.
Configuration items must be retrievable. Off-site storage for physical items is fine and recommended in potentially dangerous situations, but, if needed to restore functionality, the items must be quickly retrievable.
• Authorization. Use must be authorized and clearly scoped. Configuration management programs must have rules to ensure authorized deployment of items, which is usually done by requiring signatures of stakeholders such as managers and QA. The deployments should be strictly defined so that it is known where everything is and when its use began and ended.
• Accountability. When and where do we use it, and how long do we need to keep it? Configuration management should be able to answer questions such as “What versions were we running on this piece of equipment three years ago?” In addition, configuration management should maintain an inventory in compliance with company record retention requirements. When items are no longer needed, they should be properly discarded.
TESTING CHANGES
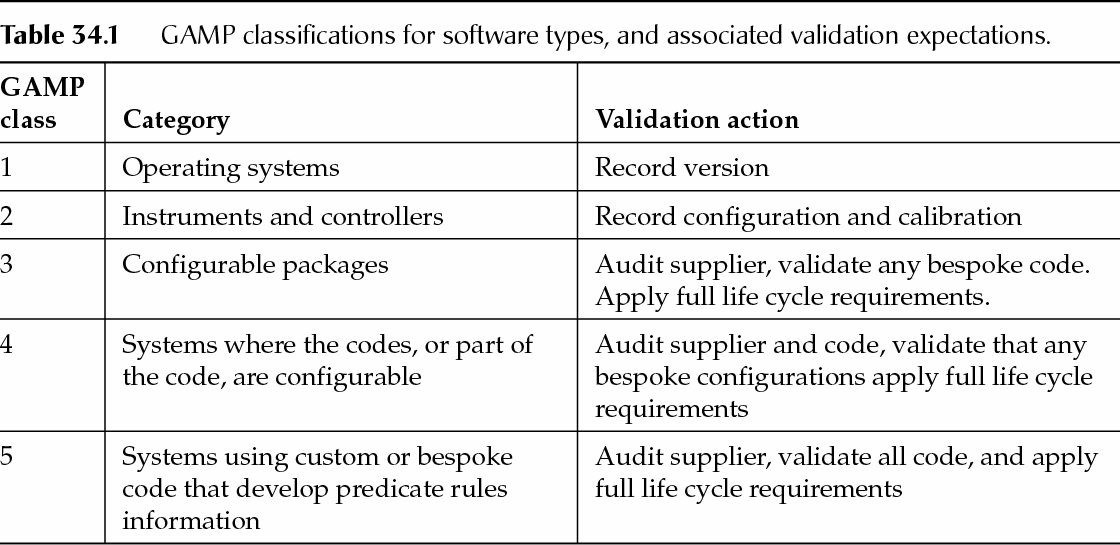
When considering testing needed relative to modifications made to a computerized system, it is helpful to categorize the type of software and what the appropriate level of testing should be. Testing at some level must be conducted to ensure that the system does what it is intended to do (that is, that it is in control). This process is referred to as maintaining the validated state. A system that has been formally tested and accepted for use should maintain that original level of reliable functionality even as it undergoes change. In this regard it is useful to be familiar with the GAMP classifications for software types and their associated validation expectations (Table 34.1).
The characteristics mentioned in regulatory requirements as being important to ensuring that automated systems are in control include access control, data protection, change control, data backup and protection, and audit trail.
Two major aspects of access control are physical and logical access. Physical access is meant to limit personnel from restricted areas (that is, doors, locks, signs). Physical restrictions comprise legitimacy checks and should include training considerations (that is, should people who do not know how to properly use the equipment be allowed access). Logical access refers to the restrictions placed on computerized systems usually in the form of identifiers, passwords, biometric restrictors, tokens, and so on. Identification restrictions apply, such as having employees have a unique personnel identifier, and password rules such as not sharing passwords and changing passwords on a regular basis. Biometric methods such as fingerprint and retinal scanners might be used. Logical controls are used to limit access to system functionality and to data, and to restrict system modification.
In general, data protection refers a system’s ability to do a number things (that is, gather, process, and transfer data accurately, maintain data integrity across interfaces, protect data such that it is beyond compromise or unauthorized modification, and retrieve data accurately). Data that can be modified should be subject to audit trails, and the audit trails must travel with the data (the data and their metadata must be linked). Importantly, data that are of regulatory interest and are kept electronically must be able to survive technological changes. It might not be useful to have batch records stored on floppy disks as not all systems are capable of handling that type of physical input. Data protection includes being able to recover data quickly regardless of its media or format.
conditions when storing media.
Backups are created as duplicate files (direct copies of files) kept in case of system failures or other disasters. Using backups to recover files and data is referred to as restoration. Archiving is a technique of moving data that are no longer actively used to a separate data storage device (or media) for long-term retention. Off-site storage of archived data is common. Data archives comprise older data that might be important and necessary for future reference. Archives may contain data that would be kept for regulatory compliance or needed to support business continuity. Again, these data must be retrievable, regardless of their format.
A system has four major components—hardware, software, documentation, and personnel—and four aspects of system maintenance. Change control (for software and documentation) and training (for personnel) are important aspects of maintenance in general, but hardware has some special considerations. Of these four categories, hardware is the most prone to physical erosion. Hardware must be maintained and calibrated to make certain that it functions as expected. Calibration is the act of periodically checking, setting, or otherwise adjusting a device to a specified operational level. Suffice to say that instruments, devices, and other hardware components used in automated systems must be calibrated. Maintenance can be thought of as three types—reactive (fix it when it breaks or degrades), preventive (change the part, usually on a periodic basis, before it breaks or degrades), or predictive (monitor the part and change it when it begins to show out-of-trend behavior).
An audit trail in terms of automated or computerized systems is an electronic accounting of who has accessed a system, when it was accessed, and what changes have been made. The audit trail often exists as a separate file but must be capable of being linked to the file that it describes. The integrity of the audit trail must be maintained as strictly as an actual system file. Keeping an accurate audit trail is an important feature of Part 11 compliance.
Periodic system monitoring may refer to many aspects of the system or the system life cycle. It can broadly refer to access monitoring (that is, who is using the system), resource monitoring (that is, how effectively is electronic traffic handled during use), assessing the state of the system (that is, are instruments properly calibrated), or checking the integrity of the system (that is, have changes had negative impact on the process or the products produced by the process). Relative to change control, periodic reviews are recommended when a system has undergone a number of changes over time. Each change may have maintained the integrity of the system, but unintended interactions between the changes may affect the overall operation of the system. In this case, a revalidation, rerunning the protocols (or portions thereof), would be used to confirm that the system remains in the validated state. Periodic reviews of unchanged operating procedures are common.


.webp)

%20Web%20of%20pharma%20.webp)


%20webofpharma.JPG)




{kind=link}